
📚 この記事について:「scRNA-seq解析 実践シリーズ(前処理編)」の記事です。全体像は「前処理の全体像とツール選択」を参照。本記事では、データの読み込みと「使えない細胞をはじく(QC)」を扱います。コードは初めてでも追えるよう、各ステップで「何をしているか」を先に説明します。
🔜 次のステップ:サンプル統合(CPU版「Harmony」/GPU版「scVI」のどちらかを選択)
前提:Scanpy が動く環境/Python の基本。新しい用語はその都度説明します。
この記事のゴール
機械から出てきたデータを読み込み、「使えない細胞」をはじいて、きれいなデータ(AnnData:scanpy が使うデータ形式)を作るところまで。
🩺 たとえると:細胞の「健康診断」です。3つの検査項目で1細胞ずつチェックし、明らかにダメなもの(壊れている・空っぽ・2細胞が混ざっている)をはじきます。この健康診断をサボると、後の解析で「存在しない細胞集団」が生まれたり、本物の違いが隠れたりします。
進め方はこの5ステップです。
- データを読み込む
- 健康診断の「3つの指標」を計算する
- 指標をグラフにして、データのクセを見る
- ダメな細胞をはじく(2つのやり方を比較)
- ノイズになる遺伝子を除き、サンプルを1つにまとめる
💡 なぜ QC(品質管理)を最初にやるの? シングルセル RNA-seq(scRNA-seq、single-cell RNA sequencing:1細胞ずつ遺伝子を測る技術)では、データ上の1つの「ドロップレット」が、必ずしも1個の元気な細胞とは限りません。空っぽの液滴、2細胞が混ざった液滴、死にかけの細胞などが紛れ込みます。 📦 宅配の仕分けにたとえると:箱(液滴)の中身が、空だったり・2個入っていたり・壊れていたりするのと同じ。だから**最初に検品(QC)**して、ちゃんとした箱だけ通します(Heumos et al., 2023)。
0. そもそもデータはどんな形?
Cell Ranger という前処理ソフトを通すと、「細胞 × 遺伝子」の巨大な表(カウント行列)が得られます。各マスは「その細胞で、その遺伝子が何分子検出されたか」という数字です。

ここで知っておきたい言葉が2つ。
- バーコード(barcode):液滴ごとに付く固有の名札。同じ名札のデータが「1細胞分」としてまとめられます。ただし前述のとおり、名札1つ=元気な細胞1個、とは限りません(名札が空席に落ちている=空液滴、2人で1枚を共有=ダブレット、のようなことが起きる)。
- UMI(unique molecular identifier):mRNA を1分子ずつ重複なく数えるための**「分子の通し番号」**。要は「その遺伝子が何分子あったか」を正確に数える仕組みです。
つまり QC とは、この**「名札と本物の細胞のズレ」を統計的に正す**作業だと考えると分かりやすいです。
1. 準備:ライブラリの読み込み
まずは使う道具(ライブラリ)を読み込みます。下のコードはそのまま実行すればOKです。
import numpy as np
import scanpy as sc
import anndata as ad
from scipy.stats import median_abs_deviation
import matplotlib.pyplot as plt
# 表示設定
sc.settings.verbosity = 1 # 0=errors, 1=warnings, 2=info, 3=hints
sc.settings.set_figure_params(dpi=80, facecolor="white")
用語:
AnnData(annotated data)は Scanpy が使うデータ箱です。発現の表(.X)に加えて、細胞ごとの情報(.obs)・遺伝子ごとの情報(.var)・計算結果(.obsm,.layers)を、1つにまとめて持ち運べるのが便利な点です。
2. データを読み込む
2-1. まずは1サンプルを読む
Cell Ranger の出力フォルダ(matrix.mtx.gz・barcodes.tsv.gz・features.tsv.gz の3点が入ったフォルダ)を指定するだけです。
adata = sc.read_10x_mtx(
"path/to/filtered_feature_bc_matrix/", # ← 自分の出力フォルダに置き換える
var_names="gene_symbols", # 遺伝子シンボルを名前に
cache=True, # 2回目以降の読み込みを高速化
)
print(adata)
2-2. 複数サンプルをまとめて扱う(テンプレート)
実験ではふつう複数サンプル(例:対照群×2、処理群×2)を扱います。サンプルの定義を1か所にまとめておくと、後で増減しても1行直すだけで済みます。
# (サンプルのラベル, 10x 出力ディレクトリ, 群ラベル)
SAMPLES = [
("Ctrl1", "path/to/ctrl1/filtered_feature_bc_matrix/", "Ctrl"),
("Ctrl2", "path/to/ctrl2/filtered_feature_bc_matrix/", "Ctrl"),
("Treat1", "path/to/treat1/filtered_feature_bc_matrix/", "Treat"),
("Treat2", "path/to/treat2/filtered_feature_bc_matrix/", "Treat"),
]
adatas = {}
for label, path, group in SAMPLES:
a = sc.read_10x_mtx(path, var_names="gene_symbols", cache=True)
a.var_names_make_unique() # 重複する遺伝子名をユニーク化
a.obs["group"] = group # 群ラベルを付与
adatas[label] = a
print(f"[{label}] {a.n_obs} cells × {a.n_vars} genes")
📁 【オプション】RNA velocity をやるなら、ここで loom を取り込む RNA velocity 解析には spliced / unspliced(成熟 / 未成熟 mRNA)のカウントが必要で、これは velocyto や kallisto|bustools が出力する loom ファイルに入っています。これをこの読み込み段階で AnnData に結合しておくのがポイントです。spliced / unspliced は
layersに格納されるため、この後の QC・ダブレット除去・遺伝子フィルタを通すたびに自動で同じ細胞・遺伝子へ絞られます。逆に前処理が終わってからマージしようとすると、生き残ったバーコードに loom を手動で対応させる必要があり、形式の不一致で事故りやすくなります。RNA velocity をやらないなら、この節は飛ばして構いません。import scvelo as scv ldata = scv.read("path/to/sample.loom", cache=True) scv.utils.clean_obs_names(adata) # バーコード名を正規化(形式の不一致対策) scv.utils.clean_obs_names(ldata) adata = scv.utils.merge(adata, ldata) # spliced / unspliced が layers に入る⚠️ 複数サンプルのとき:loom と 10x のバーコード形式が違うことが多いので、サンプルごとにマージしてから結合し、サンプル接頭辞を揃えてください。
📝 loom ファイルの作り方(velocyto / kallisto)と RNA velocity 解析の実際は、別記事で詳しく解説します。 本記事では「読み込み段階で取り込んでおける」ことだけ押さえれば十分です。
3. 健康診断の「3つの指標」(QCで何を見るか)
QC では、細胞ごとに次の3つの数字を計算します。これは健康診断の検査項目のようなもの。それぞれが何を意味し、外れているとき何を疑うかを押さえておくと、数字を機械的に当てはめずに済みます。
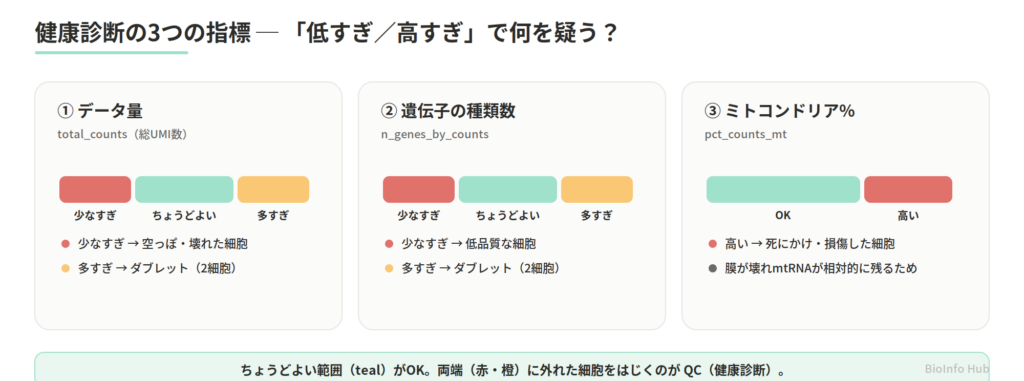
| 指標(検査項目) | 何を見る | 外れているとき疑うもの |
|---|---|---|
total_counts | データ量(総 UMI 数) | 少なすぎ=空っぽ・壊れた細胞/多すぎ=2細胞(ダブレット)の疑い |
n_genes_by_counts | 検出された遺伝子の種類数 | 少なすぎ=低品質/多すぎ=ダブレットの疑い |
pct_counts_mt | ミトコンドリア遺伝子の割合(%) | 高い=死にかけ・損傷した細胞 |
💡 なぜミトコンドリアの割合で「死細胞」が分かるの? 細胞膜が壊れると、細胞の中身(mRNA)は液滴の外へ漏れ出します。でもミトコンドリアの中の mRNA は、内膜に守られて残りやすい。結果、死にかけの細胞ほど「全体に占めるミトコンドリア mRNA の割合」が高くなります。だからこの割合が、品質の目印になります。
⚠️ ミトコンドリア遺伝子の頭文字は「種」で違う(要注意)
- ヒト:
MT-(大文字)- マウス:
mt-(小文字)ここを間違えると、ミトコンドリア割合が全細胞でゼロになり、死細胞をはじけません。最初に必ず確認しましょう。
4. 指標を計算して、まず「目で見る」
下のコードで3つの指標を一度に計算します。いきなり閾値を決めず、まずグラフで分布を眺めるのがコツです。
# 例として1サンプルで進める(実運用ではサンプルごとにループ)
adata = adatas["Ctrl1"]
# ミトコンドリア遺伝子に印をつける(マウスは "mt-"、ヒトは "MT-")
adata.var["mt"] = adata.var_names.str.startswith("mt-")
# QC 指標を計算
sc.pp.calculate_qc_metrics(
adata,
qc_vars=["mt"],
percent_top=[20], # 発現上位20遺伝子が占める割合 → 「中身の偏り」の指標
log1p=True,
inplace=True,
)
計算できたら、分布をグラフにします。
# バイオリンプロット:3指標の分布を確認
sc.pl.violin(
adata,
["n_genes_by_counts", "total_counts", "pct_counts_mt"],
jitter=0.4, multi_panel=True,
)
# 散布図:「総カウント vs 遺伝子数」を、色でミトコンドリア%を表示
sc.pl.scatter(adata, x="total_counts", y="n_genes_by_counts", color="pct_counts_mt")
💡 散布図を見ると何が分かる? ふつう「総カウント」と「遺伝子数」はきれいに比例します。この直線から右下に外れる点(カウントは多いのに遺伝子の種類が少ない)は、少数の遺伝子だけが異常に多い=技術的なゴミやダブレットの候補。1つずつ見るより、2つの関係で見たほうが外れ者が浮かび上がります。
5. ダメな細胞をはじく:2つのやり方(固定閾値 vs MAD)
ここがこの記事の核心です。フィルタの決め方には2つの流派があり、違いを知っておくと再現性が上がります。ただどっちが正しいとかはなく、データによって変わるし、ぶっちゃけQC後の細胞種アノテーションでしっかりと想定された細胞種が取れていれば問題ないと思います。逆にクラスタリングや細胞種アノテーションがうまくいっていないと思うのであれば何度かパラメーターを変えて計算してみることを勧めます。
🎓 テストの採点でたとえると
- 固定閾値=「60点未満は不合格」と点数を決め打ちするやり方。手軽だが、テストの難易度が違うと不公平。
- MAD ベース=「集団の中で大きく外れた人だけをはじく」偏差値のようなやり方。サンプルごとに事情が違っても公平に判定できる。
5-1. 固定閾値(手軽だけど根拠が弱い)
「mt% > 5% は除く」「遺伝子数 200〜6000 だけ残す」のように数値を手で決める方法です。
# 固定閾値の例(あくまで例。データごとに分布を見て決める必要がある)
adata_hard = adata[
(adata.obs.n_genes_by_counts > 200)
& (adata.obs.n_genes_by_counts < 6000)
& (adata.obs.pct_counts_mt < 5)
].copy()
print(f"固定閾値: {adata.n_obs} -> {adata_hard.n_obs} cells")
⚠️ 固定閾値の弱点
- 「mt% > 5% で切る」に根拠がない(組織や実験で適正値は変わる)。
- サンプル差を無視:同じ5%でも、Aには緩すぎ・Bには厳しすぎ、が起こる。
- 結果、人やサンプルによって再現できないフィルタになりがち。
5-2. MAD ベース(データに決めさせる・推奨)★
そこで今のおすすめは、分布から外れ値を自動で見つける MAD(中央値絶対偏差、median absolute deviation)法です(Heumos et al., 2023)。考え方は「真ん中(中央値)から大きく外れた細胞を外れ値とみなす」だけ。
def is_outlier(adata, metric: str, nmads: int = 5):
"""指定した QC 指標について、中央値 ± nmads×MAD の外を外れ値とする。"""
M = adata.obs[metric]
med = np.median(M)
mad = median_abs_deviation(M)
return (M < med - nmads * mad) | (med + nmads * mad < M)
# 一般的な指標は緩め(5 MAD)、mt% は影響が大きいので厳しめ(3 MAD)に
adata.obs["outlier"] = (
is_outlier(adata, "log1p_total_counts", 5)
| is_outlier(adata, "log1p_n_genes_by_counts", 5)
| is_outlier(adata, "pct_counts_in_top_20_genes", 5)
)
adata.obs["mt_outlier"] = is_outlier(adata, "pct_counts_mt", 3) | (adata.obs.pct_counts_mt > 8)
print("品質外れ値:", int(adata.obs.outlier.sum()))
print("mt 外れ値:", int(adata.obs.mt_outlier.sum()))
# 外れ値を除く
n_before = adata.n_obs
adata = adata[(~adata.obs.outlier) & (~adata.obs.mt_outlier)].copy()
print(f"MAD ベース: {n_before} -> {adata.n_obs} cells")
💡 なぜ「中央値」と「MAD」なの?(平均ではなく) 平均と標準偏差は、少数の極端な細胞に引っ張られてずれてしまいます。一方、中央値と MAD は極端な値に強いので、「データ自身が示す正常範囲」を素直に表せます。 また、カウント系の分布は右に大きく偏っているため、
log1p(対数)を取ってから判定します(こうすると左右のバランスが整い、MAD がうまく働きます)。nmadsは厳しさのつまみで、3=厳しめ、5=標準。最後は必ずグラフで「狙いどおり裾だけ落ちたか」を目で確認します。
📌 結局どっちを使う?
- 探索の初期や、分布がきれいで根拠ある数値が分かっている → 固定閾値でもOK
- 複数サンプル・再現性を重視・客観的に決めたい → MAD ベースが推奨
- 迷ったら、MAD で外れ値を可視化し、固定閾値と見比べて納得してから決める
6. 遺伝子のフィルタ+サンプルをまとめる
細胞のフィルタが終わったら、ごく少数の細胞でしか出てこない遺伝子を除きます(ほぼノイズで、計算も重くなるため)。
sc.pp.filter_genes(adata, min_cells=3) # 3細胞未満でしか出ない遺伝子を除外
print("遺伝子フィルタ後:", adata.shape)
最後に、QC 済みの各サンプルを1つの AnnData にまとめます。
# 各サンプルに QC を適用済みとして、まとめて結合する
combined = ad.concat(
adatas, # {label: AnnData} の辞書
label="sample", # どのサンプル由来かを obs["sample"] に記録
join="outer", # 遺伝子セットの和集合を取る
index_unique="-", # バーコード衝突を防ぐためサンプル名を付加
)
print(combined)
🔧 更新メモ:古いコードでは
adata.concatenate(...)がよく使われますが、これは非推奨です。今はanndata.concat(...)を使います(joinやindex_uniqueの挙動が明示的で、バグを避けやすい)。
7. QC は「はじく」だけじゃない(発展)
ここまでは「明らかにダメな細胞を除く」基本の QC でした。より厳密にやるなら、次の2つも検討します(本シリーズでは別記事で解説)。
⚠️ 発展的な QC(後続記事で解説)
- アンビエント RNA の除去:液滴の溶液中に漂う遊離 mRNA が、本来の発現に混ざります。
SoupX/CellBender/decontXで補正します。Python で完結させるなら CellBender が第一候補です(📖 詳しくは「CellBenderとは?」記事)。- ダブレット(2細胞)の検出:
scrubletや、scVI(single-cell variational inference)ベースのSOLOで検出・除去します。SOLO は scVI を土台にするため、統合(次の記事)で scVI を学んだ後に扱います。
まとめ
- scRNA-seq の「細胞」はノイズ(空液滴・ダブレット・死細胞)を含むので、QC が前処理の入口。健康診断のイメージ。
- 主役は3指標:
total_counts・n_genes_by_counts・pct_counts_mt。意味を理解してから閾値を決める。 - フィルタは 固定閾値(点数決め打ち)vs MAD(偏差値で自動判定)。再現性重視なら MAD が推奨。
- 仕上げに遺伝子フィルタと
anndata.concatでまとめる。
次のステップ:サンプル統合とバッチ補正 — CPU で完結する古典手法「Harmony」と、GPU 必須の AI 手法「scVI」を、それぞれ別記事で解説します。どちらか一方を選びます。
参考文献
- Heumos, L., Schaar, A. C., Lance, C., et al. (2023). Best practices for single-cell analysis across modalities. Nature Reviews Genetics, 24, 550–572. doi:10.1038/s41576-023-00586-w
- Luecken, M. D., & Theis, F. J. (2019). Current best practices in single-cell RNA-seq analysis: a tutorial. Molecular Systems Biology, 15(6), e8746. doi:10.15252/msb.20188746
- Wolf, F. A., Angerer, P., & Theis, F. J. (2018). SCANPY: large-scale single-cell gene expression data analysis. Genome Biology, 19, 15. doi:10.1186/s13059-017-1382-0
- Single-cell best practices(オンライン書籍): https://www.sc-best-practices.org

コメント